
A new mathematical model for relative quantification in real-time RT-PCR.
Use of the real-time polymerase chain response (PCR) to amplify cDNA merchandise reverse transcribed from mRNA is on the best way to changing into a routine instrument in molecular biology to review low abundance gene expression.
Real-time PCR is simple to carry out, offers the mandatory accuracy and produces dependable in addition to fast quantification outcomes.
But correct quantification of nucleic acids requires a reproducible methodology and an ample mathematical model for information evaluation. This examine enters into the actual matters of the relative quantification in real-time RT-PCR of a goal gene transcript in comparability to a reference gene transcript.
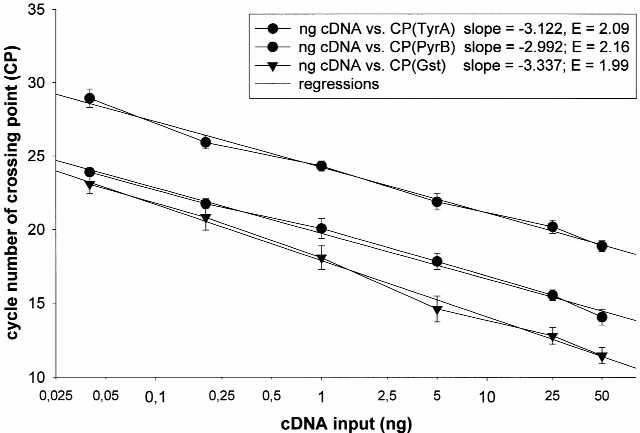
Therefore, a new mathematical model is introduced. The relative expression ratio is calculated solely from the real-time PCR efficiencies and the crossing level deviation of an unknown pattern versus a management.
This model wants no calibration curve. Control ranges have been included in the model to standardise every response run with respect to RNA integrity, pattern loading and inter-PCR variations. High accuracy and reproducibility (<2.5% variation) have been reached in LightCycler PCR utilizing the established mathematical model.
Accurate normalization of real-time quantitative RT-PCR information by geometric averaging of a number of inside management genes.
BACKGROUNDGene-expression evaluation is more and more necessary in organic analysis, with real-time reverse transcription PCR (RT-PCR) changing into the strategy of alternative for high-throughput and correct expression profiling of chosen genes.
Given the elevated sensitivity, reproducibility and enormous dynamic vary of this technique, the necessities for a correct inside management gene for normalization have develop into more and more stringent.
Although housekeeping gene expression has been reported to differ significantly, no systematic survey has correctly decided the errors associated to the frequent observe of utilizing just one management gene, nor introduced an ample approach of working round this downside.
RESULTSWe define a sturdy and revolutionary technique to establish essentially the most stably expressed management genes in a given set of tissues, and to find out the minimal variety of genes required to calculate a dependable normalization issue.
We have evaluated ten housekeeping genes from totally different abundance and purposeful lessons in varied human tissues, and demonstrated that the standard use of a single gene for normalization results in comparatively massive errors in a major proportion of samples examined.
The geometric imply of a number of rigorously chosen housekeeping genes was validated as an correct normalization issue by analyzing publicly out there microarray information.CONCLUSIONSThe normalization technique introduced here’s a prerequisite for correct RT-PCR expression profiling, which, amongst different issues, opens up the potential for finding out the organic relevance of small expression variations.